
PROTEINER OG AI
8. april 2022
Ny Machine Learning-metode giver mere meningsfulde kortlægninger af proteiner
PROTEINER
I et unikt samarbejde har forskere fra DIKU og DTU Compute skabt en teknik, der kan hjælpe biotek-industrien til hurtigere at udvikle nye proteiner. Den nye metode skal ved hjælp af kunstig intelligens og deep learning gøre det nemmere at udpege, hvilke proteiner man bør undersøge nærmere ved at kortlægge, hvordan de hænger sammen.

Biotek-industrien er konstant på jagt efter den perfekte mutation, hvor man syntetisk kombinerer egenskaber fra forskellige proteiner til at opnå en ønsket effekt. Man vil måske udvikle nye lægemidler eller enzymer, der forlænger holdbarheden på yoghurt, nedbryder plast i naturen eller får vaskepulveret til at virke effektiv ved lav vandtemperatur.
Her kan ny viden fra DTU Compute og Datalogisk Institut (DIKU) på Københavns Universitet på sigt hjælpe industrien med at speede processen op. I tidsskriftet Nature Communications forklarer forskerne, hvordan man med en ny måde at anvende Machine Learning (ML) på tegner et slags landkort over proteinerne, så man ud fra kortet kan udpege en kandidatliste af proteiner, som man skal undersøge nærmere.
– Man er de seneste år begyndt at bruge Machine Learning til at danne sig et billede af tilladte mutationer i proteiner. Problemet er dog, at man får forskellige billeder afhængig af hvilken metode, man bruger, og selv hvis man træner samme model flere gange, kan den give forskellige svar om, hvordan biologien hænger sammen. I vores arbejde kigger vi på, hvordan man kan gøre denne proces mere robust, og viser, at man derved kan udtrække væsentlig mere biologisk information, end man har kunnet tidligere. Det er et vigtigt skridt fremad for at kunne udforske mutationslandskabet i jagten efter proteiner med særlige egenskaber, siger postdoc Nicki Skafte Detlefsen fra afdelingen Kognitive Systemer på DTU Compute.
Proteinernes landkort
Et protein er en kæde af aminosyrer, og en mutation opstår, når bare én af disse aminosyrer i kæden bliver skiftet ud med en anden. Da der er 20 naturlige aminosyrer betyder det, at antallet af mutationer vokser så hurtigt, at det er fuldstændig umuligt at undersøge dem alle. Der er flere mulige mutationer, end der er atomer i universet, selv hvis man kigger på simple proteiner. Så man har ikke mulighed for eksperimentelt at afprøve alt; man er nødt til at være selektiv omkring, hvilke proteiner man vil forsøge at fremstille syntetisk.
Forskerne fra DIKU og DTU Compute har ladet deres ML-model generere et billede af, hvordan proteinerne hænger sammen. Ved at præsentere modellen for et stort antal eksempler på proteinsekvenser, lærer den at tegne et kort med en prik for hvert protein således, at tætbeslægtede proteiner placeres tæt på hinanden og fjernt beslægtede langt fra hinanden.
ML-modellen er baseret på matematik og geometri udviklet til tegning af landkort. Man skal forestille sig, at man skal lave et kort over jordkloden. Hvis man zoomer ind på Danmark, kan man godt finde ud af at tegne et kort på et stykke papir, hvor geografien er bevaret. Men hvis man skal tegne hele Jorden, laver man fejl, fordi man strækker jordkloden ud, så Arktis bliver et langt land i stedet for en pol på en kugle. Så på kortet er jordkloden skævvredet. Derfor har videnskaben inden for tegning af landkort udviklet en masse matematik, der beskriver skævvridningerne og kompenserer for det på kortet.
Det er præcis den teori, DIKU og DTU Compute har kunnet udvide til at dække Machine Learnings-modellen (Deep Learning) for proteiner. Fordi de har styr på skævvridningen på kortet, kan de også kompensere for den.
– Det gør os i stand til at tale om, hvad et fornuftigt afstandsmål er mellem proteiner, der er tæt beslægtet, og så kan vi pludselig måle det. Dermed kan vi tegne en vej gennem proteinernes landkort, der fortæller os hvilken vej, vi forventer, at et protein har udviklet sig fra til et andet - altså muteret, eftersom de alle sammen er beslægtet gennem evolutionen. På den måde kan ML-modellen måle en form for afstand mellem proteinerne og tegne optimale stier mellem lovende proteiner, siger Wouter Boomsma, lektor i sektionen for Machine Learning på DIKU.
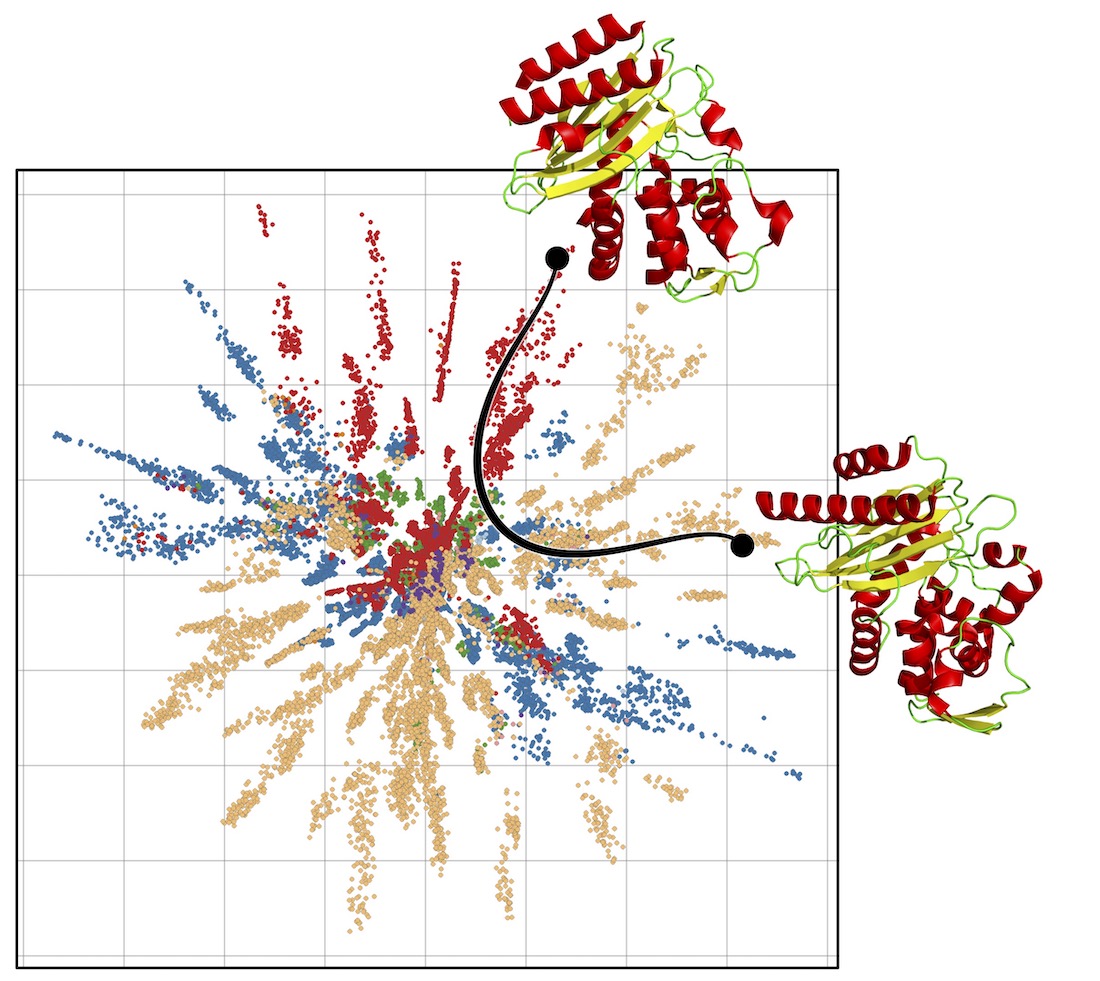
Illustration af den korteste vej mellem to proteiner, hvor man tager hensyn til afbildningens geometri. Ved at definere afstande på denne måde opnår man biologisk mere præcise og robuste konklusioner.
Forskerne har testet modellen på data for en masse proteiner, som findes ude i naturen, og hvor man kender deres struktur. Og de kan se, at afstanden mellem proteiner begynder at svare til den evolutionsmæssige udvikling af proteinerne, så proteiner, der er tæt på hinanden evolutionsmæssigt bliver placeret tæt på hinanden.
– Vi kan nu sætte to proteiner ind på landkortet og tegne kurven imellem dem. På stien mellem de to proteiner ligger mulige proteiner, som har nært beslægtede egenskaber. Det er ingen garanti, men det giver en mulighed for at have en hypotese om, hvilke proteiner det kunne være, at biotekindustrien eksperimentelt burde teste, når nye proteiner designes, siger Søren Hauberg, professor i sektionen Kognitive Systemer på DTU Compute.
Det unikke samarbejde mellem DIKU og DTU Compute blev etableret gennem et nyt center for Machine Learning in Life Science (MLLS), der startede sidste år med støtte fra Novo Nordisk Fonden. I centret samarbejder forskere i kunstig intelligens fra begge universiteter om at løse fundamentelle problemer i Machine Learning drevet af vigtige problemstillinger inden for biologien.
De udviklede protein-landkort indgår i et større projekt, der spænder fra grundforskning til industriel anvendelse, blandt andet i samarbejde med Novozymes og Novo Nordisk.
Læs mere i artiklen i Nature Communications: Learning meaningful representations of protein sequences
Kunstig intelligens, machine learning og deep learning
- Når computerprogrammer kan gøre noget ’smart’, kaldes det kunstig intelligens – ofte bare AI, som er en forkortelse af Artificial Intelligence. Kunstig intelligens er dermed et samlet begreb, der dækker over flere metoder.
- En af metoderne er Machine Learning, og den nyeste og mest avancerede brug af Machine Learning kaldes Deep Learning.
- Deep Learning er baseret på neurale netværk, som er en matematisk model, hvor modellen selv ud fra et givet datasæt og uden direkte programmering kan lære at finde mønstre i data. Fordi man anvender data, kaldes det for en datadreven model.
- I ’unsupervised learning’ er målet at træne et neuralt netværk til selv at opdage de bagvedliggende mønstre i data. Dette foregår typisk ved at forsøge at komprimere data, fordi man derigennem forkaster de tendenser i data, som er mindst hyppige, mens de væsentligste data træder mere frem, så man kan se de bagvedliggende mønstre.
- Ved hjælp af de mange gentagelser lærer netværket hvilke mønstre i data, der kan bruges til at komprimere data.
- Når modellen er trænet, testes den på ukendte data, som så også kan komprimeres til en kompakt repræsentation, der kan fortolkes til at danne videnskabelige hypoteser eller danne grundsten for andre Machine Learning-modeller.
Kontakt
Wouter Boomsma
Lektor
Datalogisk Institut
Københavns Universitet
wb@di.ku.dk
Rebekka Grage Rasmussen
Kommunikationskonsulent
Datalogisk Institut
Københavns Universitet
rgr@di.ku.dk