
Machine Learning in Biology
Modern Biology produces data in unprecedented quantities, typically of a high-dimensional nature with non-trivial correlations and substantial levels of noise. Making sense of such data is one of the main challenges of our time, not least in the context of understanding
human disease.
 Our research is concerned with developing Machine Learning algorithms in this problem domain. Currently, we focus primarily on problems in molecular biology, in particular in understanding the structure of biomolecules such as proteins. Our work is a mixture of (primarily Bayesian) Statistics, classic Bioinformatics, and Machine Learning, and we have close collaborations to research groups in each of these fields. Our probabilistic view on protein structure prediction, simulation and inference is presented in the book "Bayesian methods in structural bioinformatics" (Springer, April, 2012).
Our research is concerned with developing Machine Learning algorithms in this problem domain. Currently, we focus primarily on problems in molecular biology, in particular in understanding the structure of biomolecules such as proteins. Our work is a mixture of (primarily Bayesian) Statistics, classic Bioinformatics, and Machine Learning, and we have close collaborations to research groups in each of these fields. Our probabilistic view on protein structure prediction, simulation and inference is presented in the book "Bayesian methods in structural bioinformatics" (Springer, April, 2012).
Wouter Boomsma's research interests cover Markov chain Monte Carlo methods for protein simulations, in particular simulation under restraints from experimental data (e.g. from NMR spectroscopy), but also the problem of conducting mixed Monte Carlo / Molecular Dynamics simulations in an explicit water representation. He has worked on density estimation in various contexts, for instance for quantifying differences between molecular simulations, and as part of the development of a graphical model of the local structure of the protein backbone with the goal of speeding up molecular simulations. He has occasionally worked on strictly sequential features of molecules (i.e., the amino acid sequence), but is mostly interested in the relationship between structure and sequence. Most recently, he has initiated work towards probabilistic models of the structural environments within proteins.

A model of the local structure of proteins. See http://www.pnas.org/content/111/38/13852.abstract
Thomas Hamelryck focuses on Bayesian, probabilistic models of protein and RNA structure and their application to structure prediction, design and structure determination from experimental data (such as NMR, SAXS), including data obtained from protein ensembles. Recently, he started working on statistical evolutionary models of protein structure evolution.
Methodologically, these probabilistic models are mainly based on three key ingredients:
- Graphical models (including dynamic Bayesian networks), which are powerful machine learning methods firmly rooted in Bayesian statistics.
- Directional statistics, which is the statistics of angles, directions, orientations and other non-Euclidean data.
- The reference ratio method, which allows the formulation of probabilistic models that span multiple scales of detail. From a statistical point of view, the method makes use of a little-known version of Bayesian belief updating called "probability kinematics" or "Jeffrey's conditioning".
Graphical models, directional statistics and probability kinematics combined allow, for the first time, the formulation of valid probabilistic models of protein structure in continuous space, and with atomic detail. The same approach also allows Bayesian inference of protein ensembles (for "dynamic biomolecules" that undergo movements).
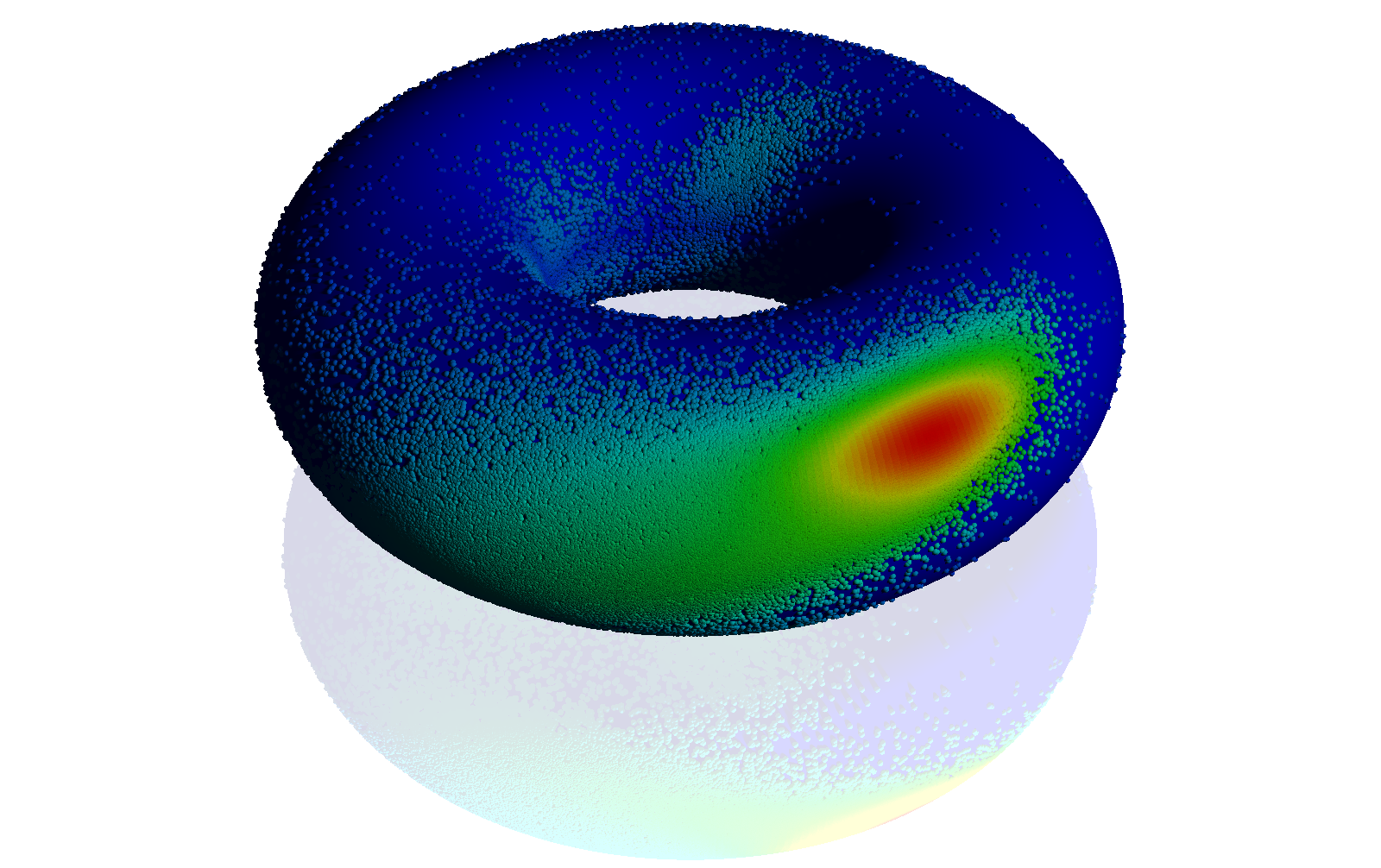
Example of a distribution on the torus: the Ramachandran distribution of backbone angles in a protein. See http://www.pnas.org/content/105/26/8932
People
| Name | Title | Phone | |
|---|---|---|---|
| Boomsma, Wouter | Professor | +4551923600 | |
| Hamelryck, Thomas Wim | Professor | +4523960613 |
Contact
 Wouter Krogh Boomsma
Wouter Krogh Boomsma
Professor
wb@di.ku.dk
Contact
 Thomas Wim Hamelryck
Thomas Wim Hamelryck
Associate Professor
thamelry@bio.ku.dk